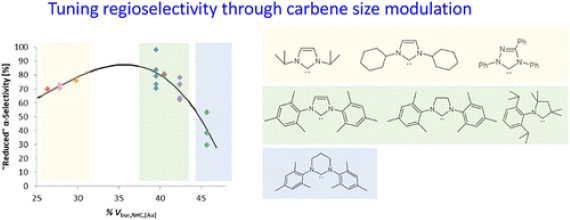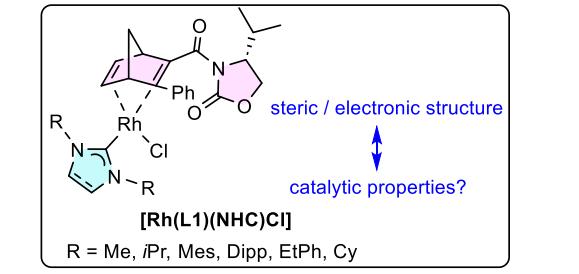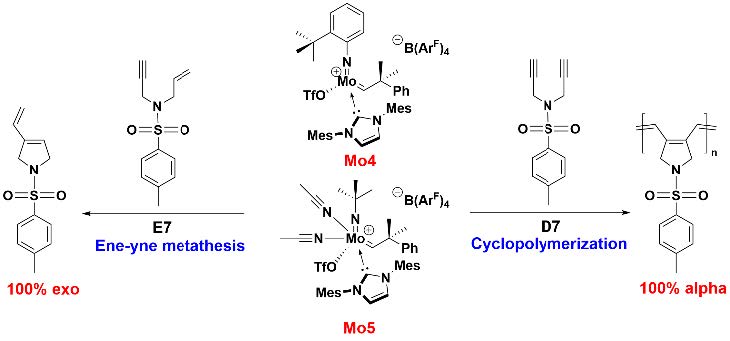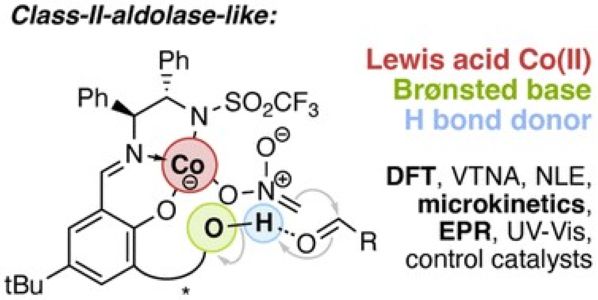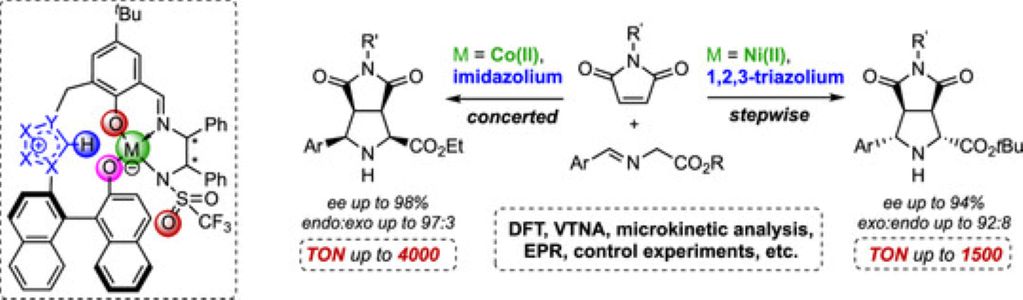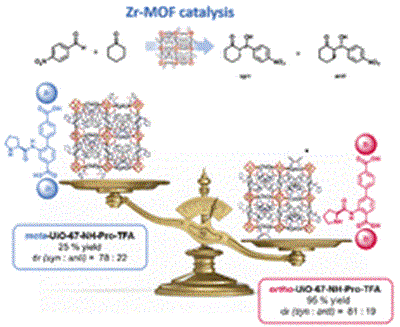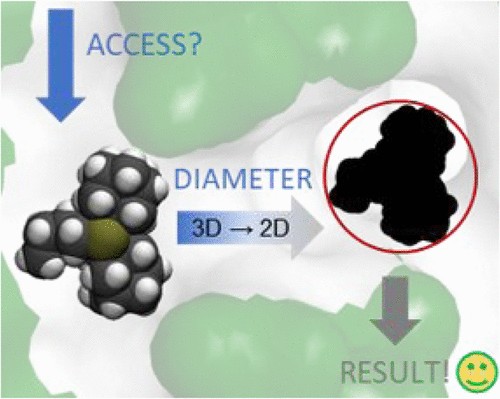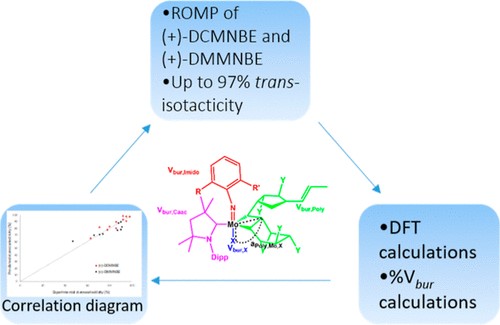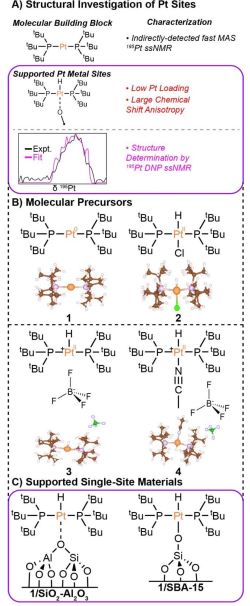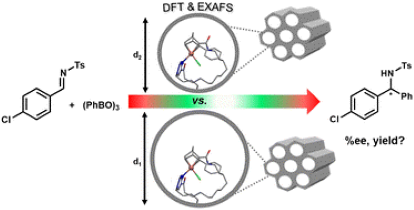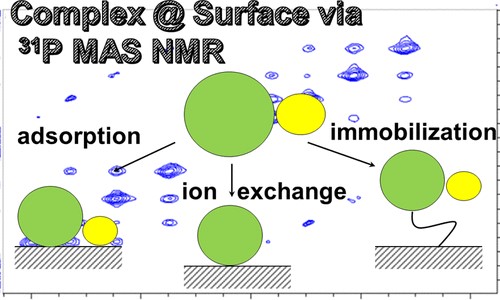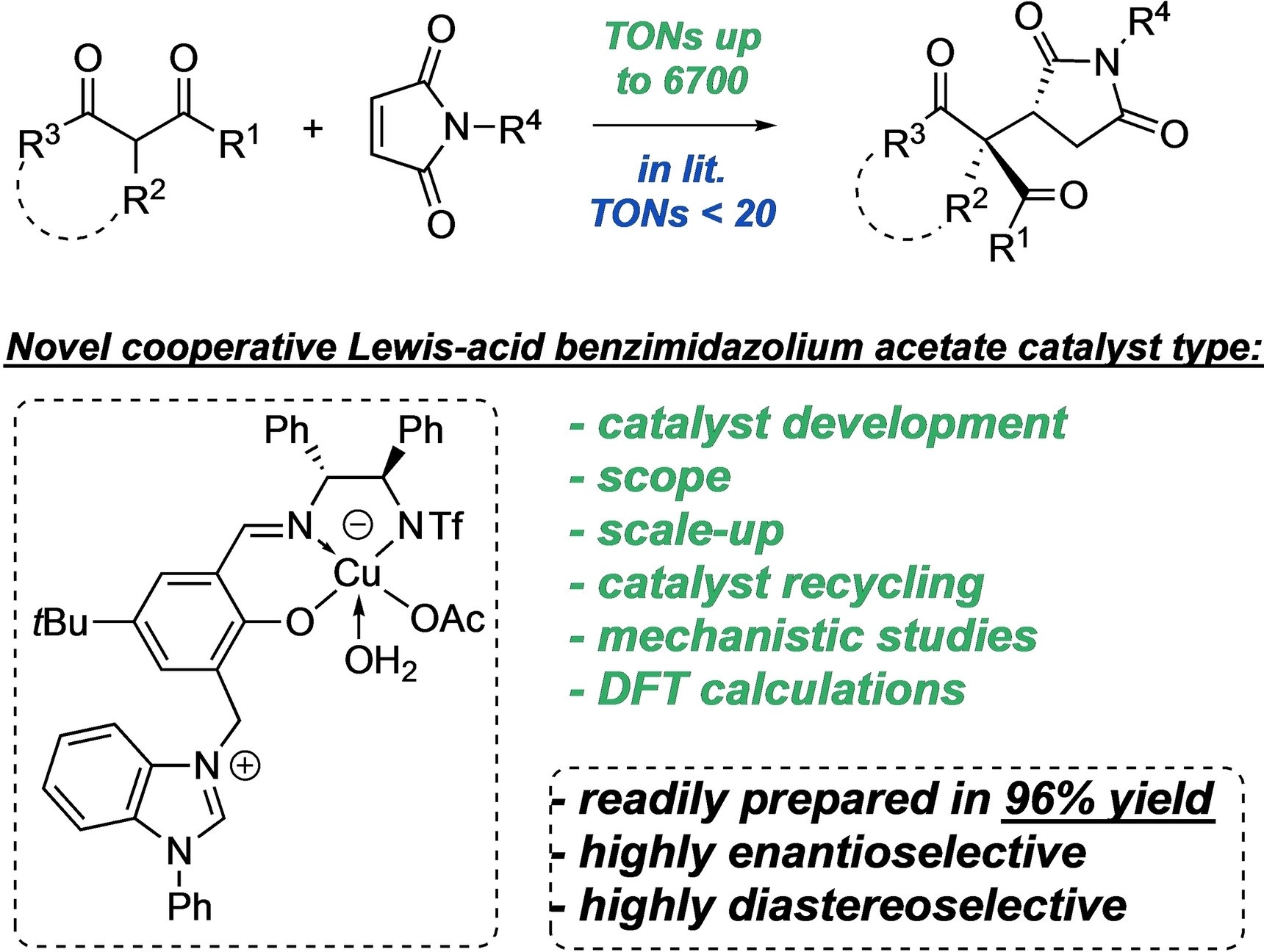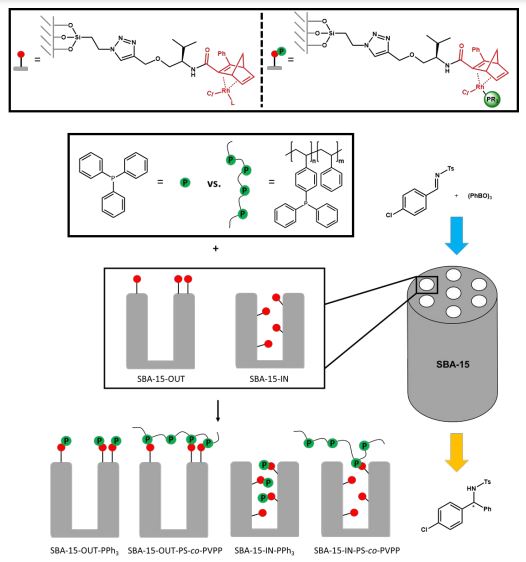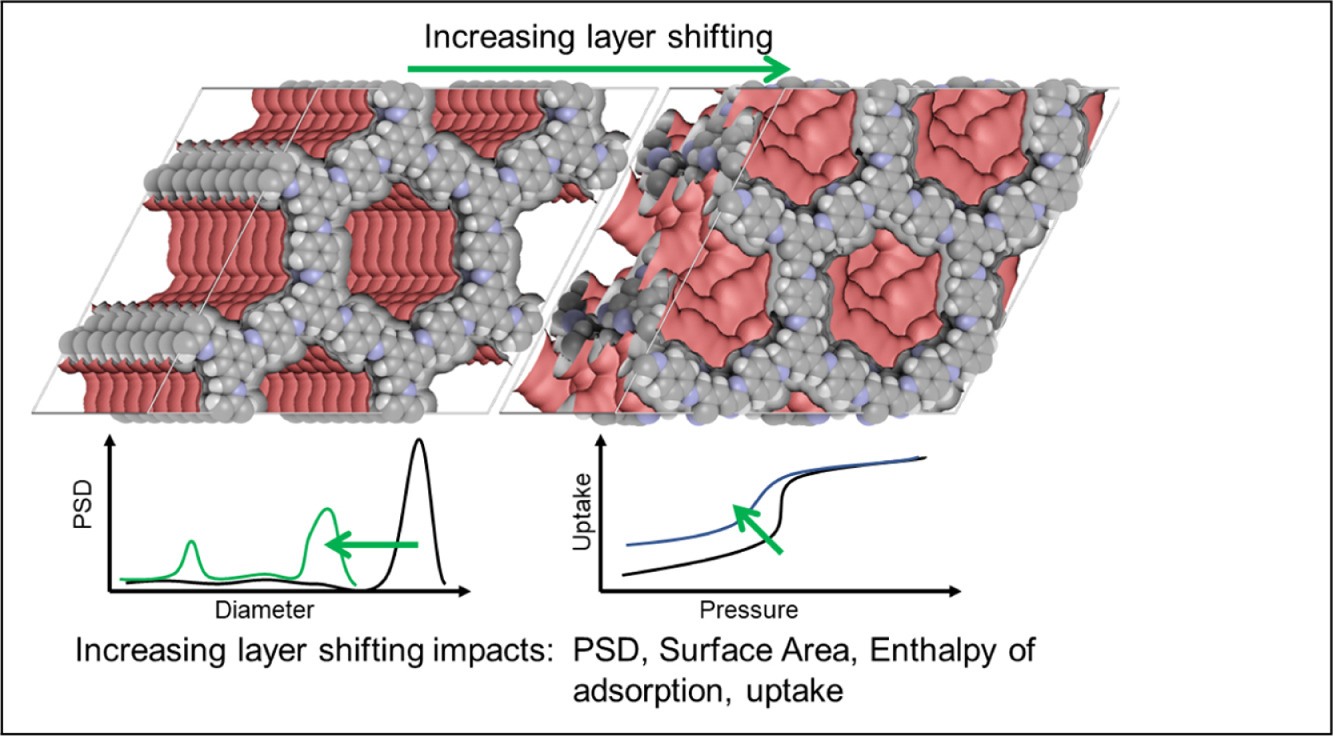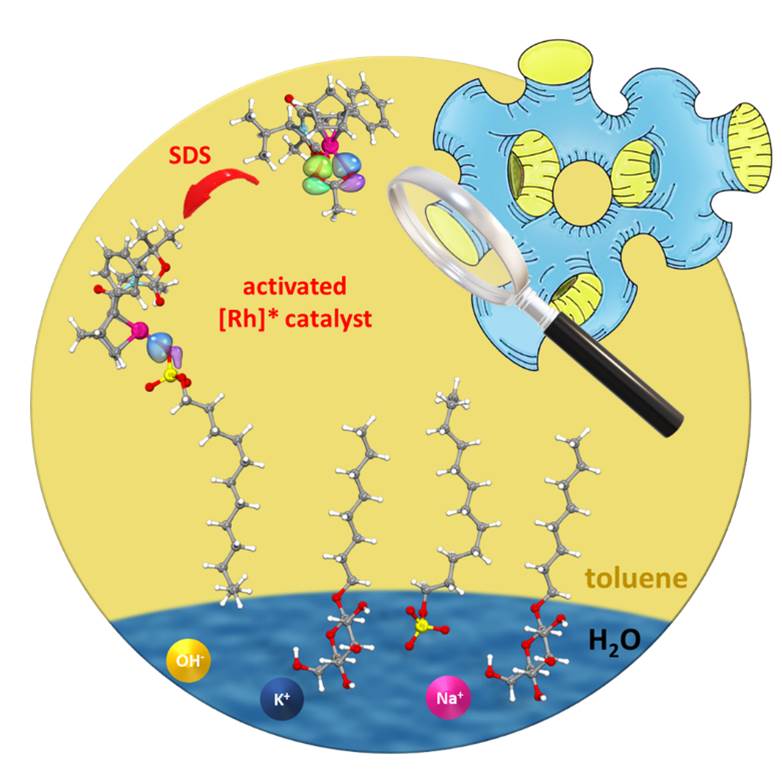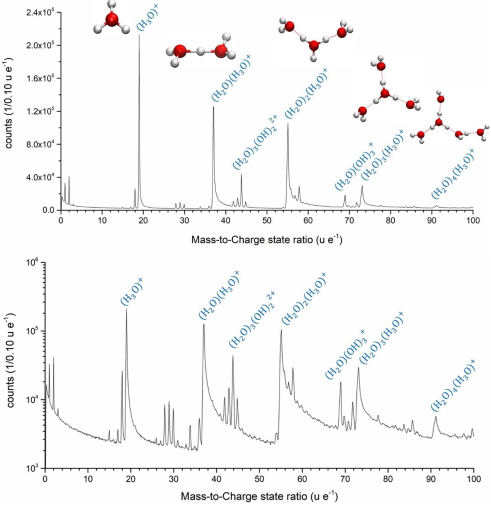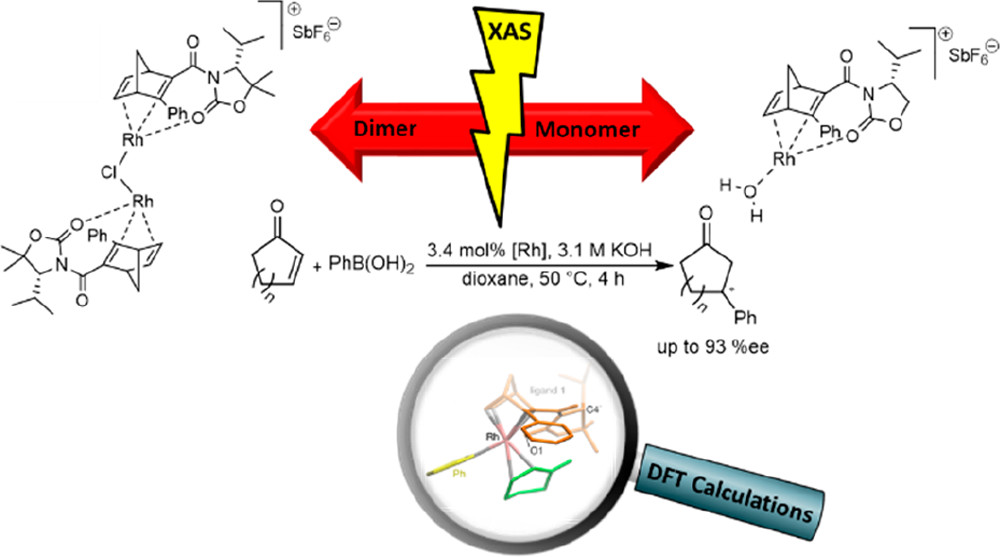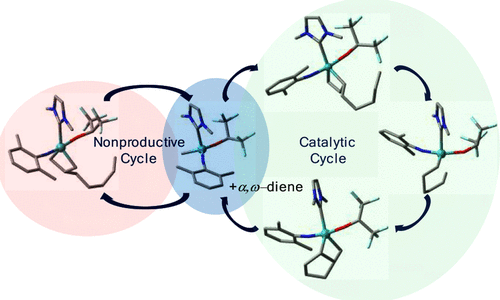
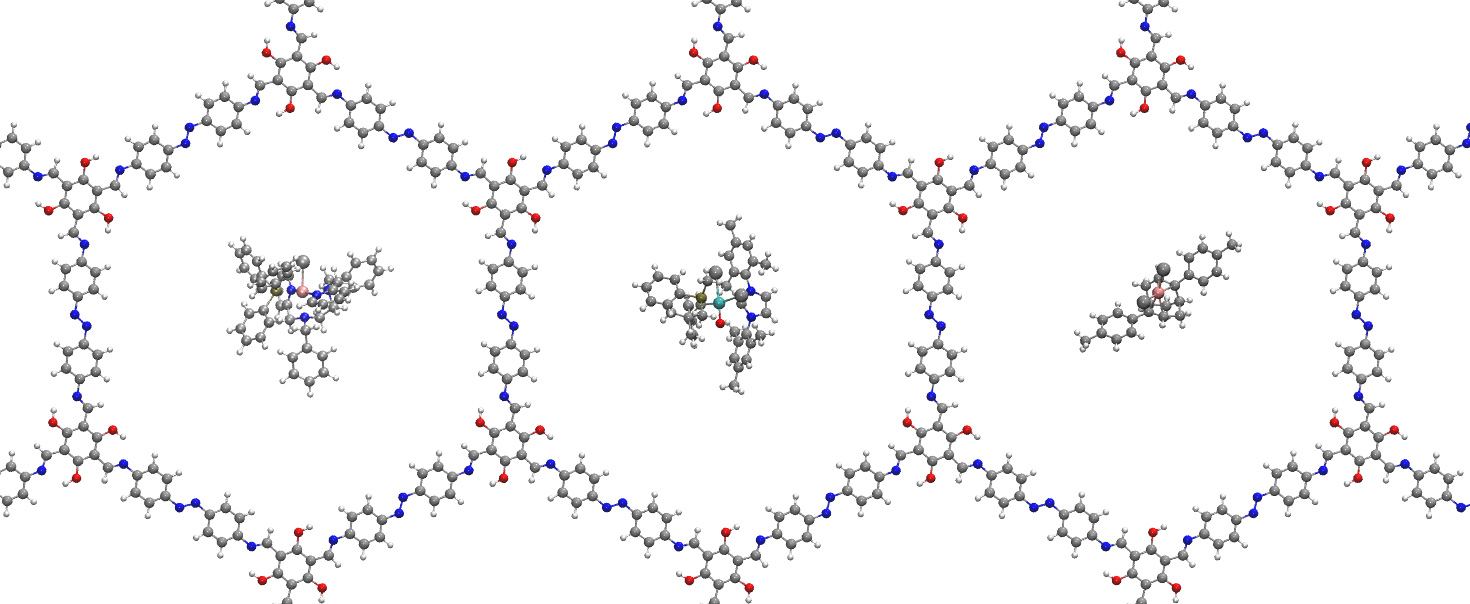
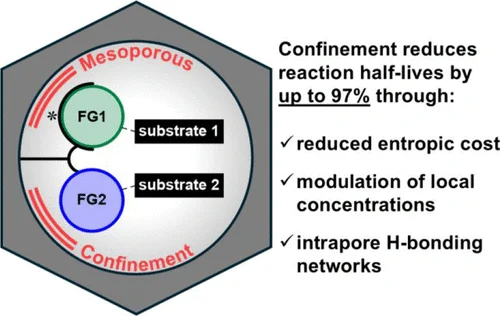
Mesoporous Confinement Enables Activity Boost in Cooperative Asymmetric Catalysis in Analogy to Enzymes
L. Rautenbach, M. Nandeshwar, E. Goldstein, M. Högler, M. Häußler, A. Allgaier, A. Bauer, D. Hornung, A. Beck, C. Franke, J. Osterbrink, J. R. Bruckner, J. Kästner, J. van Slageren, J. Pleiss, N. Hansen, M. R. Buchmeiser and R. Peters
ACS Catal 2026, 16, 11, 10371–10384.